Chapter 5 Read Counting
last updated: 2023-10-27
As usual, make sure we have the right packages for this exercise
if (!require("pacman")) install.packages("pacman"); library(pacman)
# let's load all of the files we were using and want to have again today
p_load("tidyverse", "knitr", "readr",
"pander", "BiocManager",
"dplyr", "stringr")
# We also need the Bioconductor packages "Rsubread" for today's activity.
p_load("Rsubread")5.1 featureCounts
We will first show how to use the `featureCounts() function in the Rsubread package to generate counts from the mapped .bam files.
5.1.1 Locate BAM files
Previously, we aligned our fastq files to the reference genome, generating BAM files. They should be in your “~/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp” folder, unless you chose a different place to store them.
# Where the bam files are located (default same as trimmed fastq file location)
bam_file_dir <- "~/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/"
# save list of all of those files with their full path
bam.files <- list.files(path = bam_file_dir,
pattern = ".subread.BAM$",
full.names = TRUE)
# make sure we see what we expect.
bam.files## [1] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_ETOH_REP1_R1.fastq.gz.subread.BAM"
## [2] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_ETOH_REP2_R1.fastq.gz.subread.BAM"
## [3] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_ETOH_REP3_R1.fastq.gz.subread.BAM"
## [4] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_ETOH_REP4_R1.fastq.gz.subread.BAM"
## [5] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_MOCK_REP1_R1.fastq.gz.subread.BAM"
## [6] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_MOCK_REP2_R1.fastq.gz.subread.BAM"
## [7] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_MOCK_REP3_R1.fastq.gz.subread.BAM"
## [8] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_MSN24_MOCK_REP4_R1.fastq.gz.subread.BAM"
## [9] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_ETOH_REP1_R1.fastq.gz.subread.BAM"
## [10] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_ETOH_REP2_R1.fastq.gz.subread.BAM"
## [11] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_ETOH_REP3_R1.fastq.gz.subread.BAM"
## [12] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_ETOH_REP4_R1.fastq.gz.subread.BAM"
## [13] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_MOCK_REP1_R1.fastq.gz.subread.BAM"
## [14] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_MOCK_REP2_R1.fastq.gz.subread.BAM"
## [15] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_MOCK_REP3_R1.fastq.gz.subread.BAM"
## [16] "/Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp//YPS606_WT_MOCK_REP4_R1.fastq.gz.subread.BAM"You should see the full paths to all 16 trimmed fastq bam files that we will be mapping to the reference genome today.
5.1.2 Retrieve the genome annotation
We currently have our raw reads mapped to the genome in the form of bam files. Before the differential expression analysis can proceed, these reads must be assigned and counted towards annotated genes. This can be achieved with functions in the Rsubread package, we all also see how to do this with Salmon.
We will use a bash code chunk to download the latest genome annotation
# Define the destination file path
REF_DIR="/Users/$USER/Desktop/Genomic_Data_Analysis/Reference"
# If this directory doesn't exist, you need to first complete the Read_Mapping.Rmd exercise.
# Define the URL of reference genome annotation (gtf)
# (latest from ensembl)
url="ftp://ftp.ensembl.org/pub/release-110/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz"
# Check if the file already exists at the destination location
if [ ! -f "$REF_DIR/Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz" ]; then
echo "Reference genome annotation not found, downloading..."
# If the file does not exist, download it using curl
curl -o "$REF_DIR/Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz" "$url"
echo "Downloading finished"
else
echo "File already exists at $REF_DIR Skipping download."
fi## File already exists at /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference Skipping download.Let’s take a look at the first few lines of the gtf file
# see the header columns with metadata starting with #! and delimited with \t
read.delim(
path.expand(
"~/Desktop/Genomic_Data_Analysis/Reference/Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz"
),
header = F,
sep = "\t",
nrows = 10
)## V1
## 1 #!genome-build R64-1-1
## 2 #!genome-version R64-1-1
## 3 #!genome-date 2011-09
## 4 #!genome-build-accession GCA_000146045.2
## 5 #!genebuild-last-updated 2018-10
## 6 IV
## 7 sgd
## 8 gene
## 9 8683
## 10 9756
## 11 .
## 12 -
## 13 .
## 14 gene_id YDL246C; gene_name SOR2; gene_source sgd; gene_biotype protein_coding;# We can also take a look at the first few entries to see the columns
read.delim(
path.expand(
"~/Desktop/Genomic_Data_Analysis/Reference/Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz"
),
header = F,
comment.char = "#",
strip.white = T,
nrows = 20 #just the first 20 lines
) ## V1 V2 V3 V4 V5 V6 V7 V8
## 1 IV sgd gene 8683 9756 . - .
## 2 IV sgd transcript 8683 9756 . - .
## 3 IV sgd exon 8683 9756 . - .
## 4 IV sgd CDS 8686 9756 . - 0
## 5 IV sgd start_codon 9754 9756 . - 0
## 6 IV sgd stop_codon 8683 8685 . - 0
## 7 IV sgd gene 17577 18566 . - .
## 8 IV sgd transcript 17577 18566 . - .
## 9 IV sgd exon 17577 18566 . - .
## 10 IV sgd CDS 17580 18566 . - 0
## 11 IV sgd start_codon 18564 18566 . - 0
## 12 IV sgd stop_codon 17577 17579 . - 0
## 13 IV sgd gene 1248154 1249821 . - .
## 14 IV sgd transcript 1248154 1249821 . - .
## 15 IV sgd exon 1248154 1249821 . - .
## 16 IV sgd CDS 1248157 1249821 . - 0
## 17 IV sgd start_codon 1249819 1249821 . - 0
## 18 IV sgd stop_codon 1248154 1248156 . - 0
## 19 IV sgd gene 289572 290081 . - .
## 20 IV sgd transcript 289572 290081 . - .
## V9
## 1 gene_id YDL246C; gene_name SOR2; gene_source sgd; gene_biotype protein_coding;
## 2 gene_id YDL246C; transcript_id YDL246C_mRNA; gene_name SOR2; gene_source sgd; gene_biotype protein_coding; transcript_name SOR2; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 3 gene_id YDL246C; transcript_id YDL246C_mRNA; exon_number 1; gene_name SOR2; gene_source sgd; gene_biotype protein_coding; transcript_name SOR2; transcript_source sgd; transcript_biotype protein_coding; exon_id YDL246C_mRNA-E1; tag Ensembl_canonical;
## 4 gene_id YDL246C; transcript_id YDL246C_mRNA; exon_number 1; gene_name SOR2; gene_source sgd; gene_biotype protein_coding; transcript_name SOR2; transcript_source sgd; transcript_biotype protein_coding; protein_id YDL246C; tag Ensembl_canonical;
## 5 gene_id YDL246C; transcript_id YDL246C_mRNA; exon_number 1; gene_name SOR2; gene_source sgd; gene_biotype protein_coding; transcript_name SOR2; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 6 gene_id YDL246C; transcript_id YDL246C_mRNA; exon_number 1; gene_name SOR2; gene_source sgd; gene_biotype protein_coding; transcript_name SOR2; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 7 gene_id YDL243C; gene_name AAD4; gene_source sgd; gene_biotype protein_coding;
## 8 gene_id YDL243C; transcript_id YDL243C_mRNA; gene_name AAD4; gene_source sgd; gene_biotype protein_coding; transcript_name AAD4; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 9 gene_id YDL243C; transcript_id YDL243C_mRNA; exon_number 1; gene_name AAD4; gene_source sgd; gene_biotype protein_coding; transcript_name AAD4; transcript_source sgd; transcript_biotype protein_coding; exon_id YDL243C_mRNA-E1; tag Ensembl_canonical;
## 10 gene_id YDL243C; transcript_id YDL243C_mRNA; exon_number 1; gene_name AAD4; gene_source sgd; gene_biotype protein_coding; transcript_name AAD4; transcript_source sgd; transcript_biotype protein_coding; protein_id YDL243C; tag Ensembl_canonical;
## 11 gene_id YDL243C; transcript_id YDL243C_mRNA; exon_number 1; gene_name AAD4; gene_source sgd; gene_biotype protein_coding; transcript_name AAD4; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 12 gene_id YDL243C; transcript_id YDL243C_mRNA; exon_number 1; gene_name AAD4; gene_source sgd; gene_biotype protein_coding; transcript_name AAD4; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 13 gene_id YDR387C; gene_name CIN10; gene_source sgd; gene_biotype protein_coding;
## 14 gene_id YDR387C; transcript_id YDR387C_mRNA; gene_name CIN10; gene_source sgd; gene_biotype protein_coding; transcript_name CIN10; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 15 gene_id YDR387C; transcript_id YDR387C_mRNA; exon_number 1; gene_name CIN10; gene_source sgd; gene_biotype protein_coding; transcript_name CIN10; transcript_source sgd; transcript_biotype protein_coding; exon_id YDR387C_mRNA-E1; tag Ensembl_canonical;
## 16 gene_id YDR387C; transcript_id YDR387C_mRNA; exon_number 1; gene_name CIN10; gene_source sgd; gene_biotype protein_coding; transcript_name CIN10; transcript_source sgd; transcript_biotype protein_coding; protein_id YDR387C; tag Ensembl_canonical;
## 17 gene_id YDR387C; transcript_id YDR387C_mRNA; exon_number 1; gene_name CIN10; gene_source sgd; gene_biotype protein_coding; transcript_name CIN10; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 18 gene_id YDR387C; transcript_id YDR387C_mRNA; exon_number 1; gene_name CIN10; gene_source sgd; gene_biotype protein_coding; transcript_name CIN10; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;
## 19 gene_id YDL094C; gene_source sgd; gene_biotype protein_coding;
## 20 gene_id YDL094C; transcript_id YDL094C_mRNA; gene_source sgd; gene_biotype protein_coding; transcript_source sgd; transcript_biotype protein_coding; tag Ensembl_canonical;There are 9 columns in a standard gtf file, information about each is available here: https://useast.ensembl.org/info/website/upload/gff.html
Note that version 2 of gff is identical to the gtf format.
5.1.3 Counting with FeatureCounts
library(Rsubread)
# Set path of the reference annotation gzipped gtf file
reference_annotation = "~/Desktop/Genomic_Data_Analysis/Reference/Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz"We can see the arguments available with the align function from the Rsubread package
## function (files, annot.inbuilt = "mm39", annot.ext = NULL, isGTFAnnotationFile = FALSE,
## GTF.featureType = "exon", GTF.attrType = "gene_id", GTF.attrType.extra = NULL,
## chrAliases = NULL, useMetaFeatures = TRUE, allowMultiOverlap = FALSE,
## minOverlap = 1, fracOverlap = 0, fracOverlapFeature = 0,
## largestOverlap = FALSE, nonOverlap = NULL, nonOverlapFeature = NULL,
## readShiftType = "upstream", readShiftSize = 0, readExtension5 = 0,
## readExtension3 = 0, read2pos = NULL, countMultiMappingReads = TRUE,
## fraction = FALSE, isLongRead = FALSE, minMQS = 0, splitOnly = FALSE,
## nonSplitOnly = FALSE, primaryOnly = FALSE, ignoreDup = FALSE,
## strandSpecific = 0, juncCounts = FALSE, genome = NULL, isPairedEnd = FALSE,
## countReadPairs = TRUE, requireBothEndsMapped = FALSE, checkFragLength = FALSE,
## minFragLength = 50, maxFragLength = 600, countChimericFragments = TRUE,
## autosort = TRUE, nthreads = 1, byReadGroup = FALSE, reportReads = NULL,
## reportReadsPath = NULL, maxMOp = 10, tmpDir = ".", verbose = FALSE)
## NULLThe Phred offset determines the encoding for the base-calling quality string in the FASTQ file. For the Illumina 1.8 format onwards, this encoding is set at +33. However, older formats may use a +64 encoding. Users should ensure that the correct encoding is specified during alignment. If unsure, one can examine the first several quality strings in the FASTQ file. A good rule of thumb is to check whether lower-case letters are present (+64 encoding) or absent (+33).
# This command counts the number of each feature per fastq file,
#. generating an output we can use later.
fc <- featureCounts(bam.files,
annot.ext = reference_annotation,
isGTFAnnotationFile = TRUE,
GTF.featureType = "exon"
)##
## ========== _____ _ _ ____ _____ ______ _____
## ===== / ____| | | | _ \| __ \| ____| /\ | __ \
## ===== | (___ | | | | |_) | |__) | |__ / \ | | | |
## ==== \___ \| | | | _ <| _ /| __| / /\ \ | | | |
## ==== ____) | |__| | |_) | | \ \| |____ / ____ \| |__| |
## ========== |_____/ \____/|____/|_| \_\______/_/ \_\_____/
## Rsubread 2.14.2
##
## //========================== featureCounts setting ===========================\\
## || ||
## || Input files : 16 BAM files ||
## || ||
## || YPS606_MSN24_ETOH_REP1_R1.fastq.gz.subread.BAM ||
## || YPS606_MSN24_ETOH_REP2_R1.fastq.gz.subread.BAM ||
## || YPS606_MSN24_ETOH_REP3_R1.fastq.gz.subread.BAM ||
## || YPS606_MSN24_ETOH_REP4_R1.fastq.gz.subread.BAM ||
## || YPS606_MSN24_MOCK_REP1_R1.fastq.gz.subread.BAM ||
## || YPS606_MSN24_MOCK_REP2_R1.fastq.gz.subread.BAM ||
## || YPS606_MSN24_MOCK_REP3_R1.fastq.gz.subread.BAM ||
## || YPS606_MSN24_MOCK_REP4_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_ETOH_REP1_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_ETOH_REP2_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_ETOH_REP3_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_ETOH_REP4_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_MOCK_REP1_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_MOCK_REP2_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_MOCK_REP3_R1.fastq.gz.subread.BAM ||
## || YPS606_WT_MOCK_REP4_R1.fastq.gz.subread.BAM ||
## || ||
## || Paired-end : no ||
## || Count read pairs : no ||
## || Annotation : Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz ... ||
## || Dir for temp files : . ||
## || Threads : 1 ||
## || Level : meta-feature level ||
## || Multimapping reads : counted ||
## || Multi-overlapping reads : not counted ||
## || Min overlapping bases : 1 ||
## || ||
## \\============================================================================//
##
## //================================= Running ==================================\\
## || ||
## || Load annotation file Saccharomyces_cerevisiae.R64-1-1.110.gtf.gz ... ||
## || Features : 7507 ||
## || Meta-features : 7127 ||
## || Chromosomes/contigs : 17 ||
## || ||
## || Process BAM file YPS606_MSN24_ETOH_REP1_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 233278 ||
## || Successfully assigned alignments : 175843 (75.4%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_MSN24_ETOH_REP2_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 215810 ||
## || Successfully assigned alignments : 161818 (75.0%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_MSN24_ETOH_REP3_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 199076 ||
## || Successfully assigned alignments : 148581 (74.6%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_MSN24_ETOH_REP4_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 205792 ||
## || Successfully assigned alignments : 153525 (74.6%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_MSN24_MOCK_REP1_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 167075 ||
## || Successfully assigned alignments : 122364 (73.2%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_MSN24_MOCK_REP2_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 169754 ||
## || Successfully assigned alignments : 126310 (74.4%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_MSN24_MOCK_REP3_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 210001 ||
## || Successfully assigned alignments : 151958 (72.4%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_MSN24_MOCK_REP4_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 208329 ||
## || Successfully assigned alignments : 153346 (73.6%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_ETOH_REP1_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 181587 ||
## || Successfully assigned alignments : 137526 (75.7%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_ETOH_REP2_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 201551 ||
## || Successfully assigned alignments : 151322 (75.1%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_ETOH_REP3_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 214745 ||
## || Successfully assigned alignments : 161909 (75.4%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_ETOH_REP4_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 187319 ||
## || Successfully assigned alignments : 141422 (75.5%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_MOCK_REP1_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 223539 ||
## || Successfully assigned alignments : 165863 (74.2%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_MOCK_REP2_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 187469 ||
## || Successfully assigned alignments : 138324 (73.8%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_MOCK_REP3_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 224767 ||
## || Successfully assigned alignments : 163337 (72.7%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Process BAM file YPS606_WT_MOCK_REP4_R1.fastq.gz.subread.BAM... ||
## || Single-end reads are included. ||
## || Total alignments : 206865 ||
## || Successfully assigned alignments : 152394 (73.7%) ||
## || Running time : 0.00 minutes ||
## || ||
## || Write the final count table. ||
## || Write the read assignment summary. ||
## || ||
## \\============================================================================//We can see what all is stored in the featureCounts output object
## [1] "counts" "annotation" "targets" "stat"The statistics of the read mapping can be seen with fc$stats. This reports the numbers of unassigned reads and the reasons why they are not assigned (eg. ambiguity, multi-mapping, secondary alignment, mapping quality, fragment length, chimera, read duplicate, non-junction and so on), in addition to the number of successfully assigned reads for each library.
## Status YPS606_MSN24_ETOH_REP1_R1.fastq.gz.subread.BAM
## 1 Assigned 175843
## 2 Unassigned_Unmapped 28887
## 3 Unassigned_Read_Type 0
## 4 Unassigned_Singleton 0
## 5 Unassigned_MappingQuality 0
## 6 Unassigned_Chimera 0
## 7 Unassigned_FragmentLength 0
## 8 Unassigned_Duplicate 0
## 9 Unassigned_MultiMapping 0
## 10 Unassigned_Secondary 0
## 11 Unassigned_NonSplit 0
## 12 Unassigned_NoFeatures 16741
## 13 Unassigned_Overlapping_Length 0
## 14 Unassigned_Ambiguity 11807
## YPS606_MSN24_ETOH_REP2_R1.fastq.gz.subread.BAM
## 1 161818
## 2 26525
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 15747
## 13 0
## 14 11720
## YPS606_MSN24_ETOH_REP3_R1.fastq.gz.subread.BAM
## 1 148581
## 2 25383
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 14166
## 13 0
## 14 10946
## YPS606_MSN24_ETOH_REP4_R1.fastq.gz.subread.BAM
## 1 153525
## 2 27007
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 13608
## 13 0
## 14 11652
## YPS606_MSN24_MOCK_REP1_R1.fastq.gz.subread.BAM
## 1 122364
## 2 23961
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 12806
## 13 0
## 14 7944
## YPS606_MSN24_MOCK_REP2_R1.fastq.gz.subread.BAM
## 1 126310
## 2 23452
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 11908
## 13 0
## 14 8084
## YPS606_MSN24_MOCK_REP3_R1.fastq.gz.subread.BAM
## 1 151958
## 2 31337
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 16741
## 13 0
## 14 9965
## YPS606_MSN24_MOCK_REP4_R1.fastq.gz.subread.BAM
## 1 153346
## 2 30580
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 14454
## 13 0
## 14 9949
## YPS606_WT_ETOH_REP1_R1.fastq.gz.subread.BAM
## 1 137526
## 2 22387
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 11511
## 13 0
## 14 10163
## YPS606_WT_ETOH_REP2_R1.fastq.gz.subread.BAM
## 1 151322
## 2 24647
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 14578
## 13 0
## 14 11004
## YPS606_WT_ETOH_REP3_R1.fastq.gz.subread.BAM
## 1 161909
## 2 26246
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 15064
## 13 0
## 14 11526
## YPS606_WT_ETOH_REP4_R1.fastq.gz.subread.BAM
## 1 141422
## 2 23167
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 12670
## 13 0
## 14 10060
## YPS606_WT_MOCK_REP1_R1.fastq.gz.subread.BAM
## 1 165863
## 2 30132
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 16586
## 13 0
## 14 10958
## YPS606_WT_MOCK_REP2_R1.fastq.gz.subread.BAM
## 1 138324
## 2 26218
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 13984
## 13 0
## 14 8943
## YPS606_WT_MOCK_REP3_R1.fastq.gz.subread.BAM
## 1 163337
## 2 32663
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 17777
## 13 0
## 14 10990
## YPS606_WT_MOCK_REP4_R1.fastq.gz.subread.BAM
## 1 152394
## 2 29335
## 3 0
## 4 0
## 5 0
## 6 0
## 7 0
## 8 0
## 9 0
## 10 0
## 11 0
## 12 14960
## 13 0
## 14 101765.1.4 Counts object
The counts for the samples are stored in fc$counts.
We can look at the dimensions of the counts to see how many genes and samples are present. The first number is the number of genes and the second number is the number of samples.
## [1] 7127 16let’s take a look at the first few lines of fc$counts
## YPS606_MSN24_ETOH_REP1_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 2
## YDR387C 6
## YDL094C 4
## YDR438W 5
## YDR523C 1
## YPS606_MSN24_ETOH_REP2_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 1
## YDR387C 10
## YDL094C 5
## YDR438W 6
## YDR523C 1
## YPS606_MSN24_ETOH_REP3_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 1
## YDR387C 7
## YDL094C 7
## YDR438W 5
## YDR523C 0
## YPS606_MSN24_ETOH_REP4_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 4
## YDR387C 6
## YDL094C 4
## YDR438W 3
## YDR523C 0
## YPS606_MSN24_MOCK_REP1_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 1
## YDR387C 1
## YDL094C 3
## YDR438W 4
## YDR523C 0
## YPS606_MSN24_MOCK_REP2_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 1
## YDR387C 3
## YDL094C 1
## YDR438W 1
## YDR523C 0
## YPS606_MSN24_MOCK_REP3_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 0
## YDR387C 3
## YDL094C 3
## YDR438W 3
## YDR523C 0
## YPS606_MSN24_MOCK_REP4_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 0
## YDR387C 6
## YDL094C 1
## YDR438W 1
## YDR523C 1
## YPS606_WT_ETOH_REP1_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 4
## YDR387C 9
## YDL094C 2
## YDR438W 1
## YDR523C 0
## YPS606_WT_ETOH_REP2_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 0
## YDR387C 7
## YDL094C 4
## YDR438W 3
## YDR523C 1
## YPS606_WT_ETOH_REP3_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 3
## YDR387C 12
## YDL094C 2
## YDR438W 4
## YDR523C 1
## YPS606_WT_ETOH_REP4_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 2
## YDR387C 7
## YDL094C 5
## YDR438W 7
## YDR523C 1
## YPS606_WT_MOCK_REP1_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 3
## YDR387C 4
## YDL094C 2
## YDR438W 1
## YDR523C 0
## YPS606_WT_MOCK_REP2_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 1
## YDR387C 2
## YDL094C 3
## YDR438W 3
## YDR523C 0
## YPS606_WT_MOCK_REP3_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 0
## YDR387C 9
## YDL094C 6
## YDR438W 2
## YDR523C 0
## YPS606_WT_MOCK_REP4_R1.fastq.gz.subread.BAM
## YDL246C 0
## YDL243C 0
## YDR387C 6
## YDL094C 5
## YDR438W 4
## YDR523C 0The row names of the fc$counts matrix represent the Systematic Name for each gene (can be Entrez gene identifiers for other organisms) and the column names are the output filenames from calling the align function.
The annotation slot shows the annotation information that featureCounts used to summarise reads over genes.
## GeneID Chr Start End Strand Length
## 1 YDL246C IV 8683 9756 - 1074
## 2 YDL243C IV 17577 18566 - 990
## 3 YDR387C IV 1248154 1249821 - 1668
## 4 YDL094C IV 289572 290081 - 510
## 5 YDR438W IV 1338274 1339386 + 1113
## 6 YDR523C IV 1485566 1487038 - 14735.1.5 Saving fc object for future use
We will need to use this object in our next class. We can use the R function saveRDS() to save the R object to your computer, so it can be accessed at a later date.
# create a directory for the count output to go into if not already present
dir_output_counts <- path.expand("~/Desktop/Genomic_Data_Analysis/Data/Counts/Rsubread/")
if (!dir.exists(dir_output_counts)) {dir.create(dir_output_counts, recursive = TRUE)}
# save the R data object
saveRDS(object = fc, file = paste0(dir_output_counts,"rsubread.yeast_fc_output.Rds"))
# often, we want to share this file as a tsv file. Here is how we can do that:
write_tsv(data.frame(
fc$annotation[,"GeneID"],
fc$counts,
stringsAsFactors=FALSE),
file=paste0(dir_output_counts,"rsubread.gene_counts.merged.yeast.tsv"))5.1.6 RSubread QC
We can have a look at the quality scores associated with each base that has been called by the sequencing machine using the qualityScores function in Rsubread.
Let’s extract quality scores for 50 reads for the fastq file .
# Extract quality scores
qs <- qualityScores(
filename="~/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_ETOH_REP1_R1.fastq.gz",
nreads=50)##
## qualityScores Rsubread 2.14.2
##
## Scan the input file...
## Totally 233278 reads were scanned; the sampling interval is 4665.
## Now extract read quality information...
##
## Completed successfully. Quality scores for 50 reads (equally spaced in the file) are returned.## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
## [1,] 32 32 37 37 37 41 41 41 37 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41
## [2,] 32 32 37 37 37 41 41 41 41 41 41 41 37 41 41 41 41 41 41 41 41 41 41 41 41
## [3,] 32 32 37 37 37 41 41 41 41 41 37 41 41 37 41 41 41 37 41 41 41 41 41 41 41
## [4,] 32 32 37 37 37 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41
## [5,] 32 32 37 37 37 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41
## [6,] 32 32 37 37 37 41 41 41 41 41 41 27 37 41 41 41 41 41 41 41 41 41 41 41 41
## 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50
## [1,] 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41
## [2,] 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41
## [3,] 41 41 41 41 41 41 41 41 41 41 41 41 41 41 37 41 41 41 41 41 41 41 41 41 41
## [4,] 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41
## [5,] 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41
## [6,] 41 41 41 41 37 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41 41We are randomly sampling 50 reads from the file and seeing the quality scores. A quality score of 30 corresponds to a 1 in 1000 chance of an incorrect base call. (A quality score of 10 is a 1 in 10 chance of an incorrect base call.) To look at the overall distribution of quality scores across the sampled reads, we can look at a boxplot
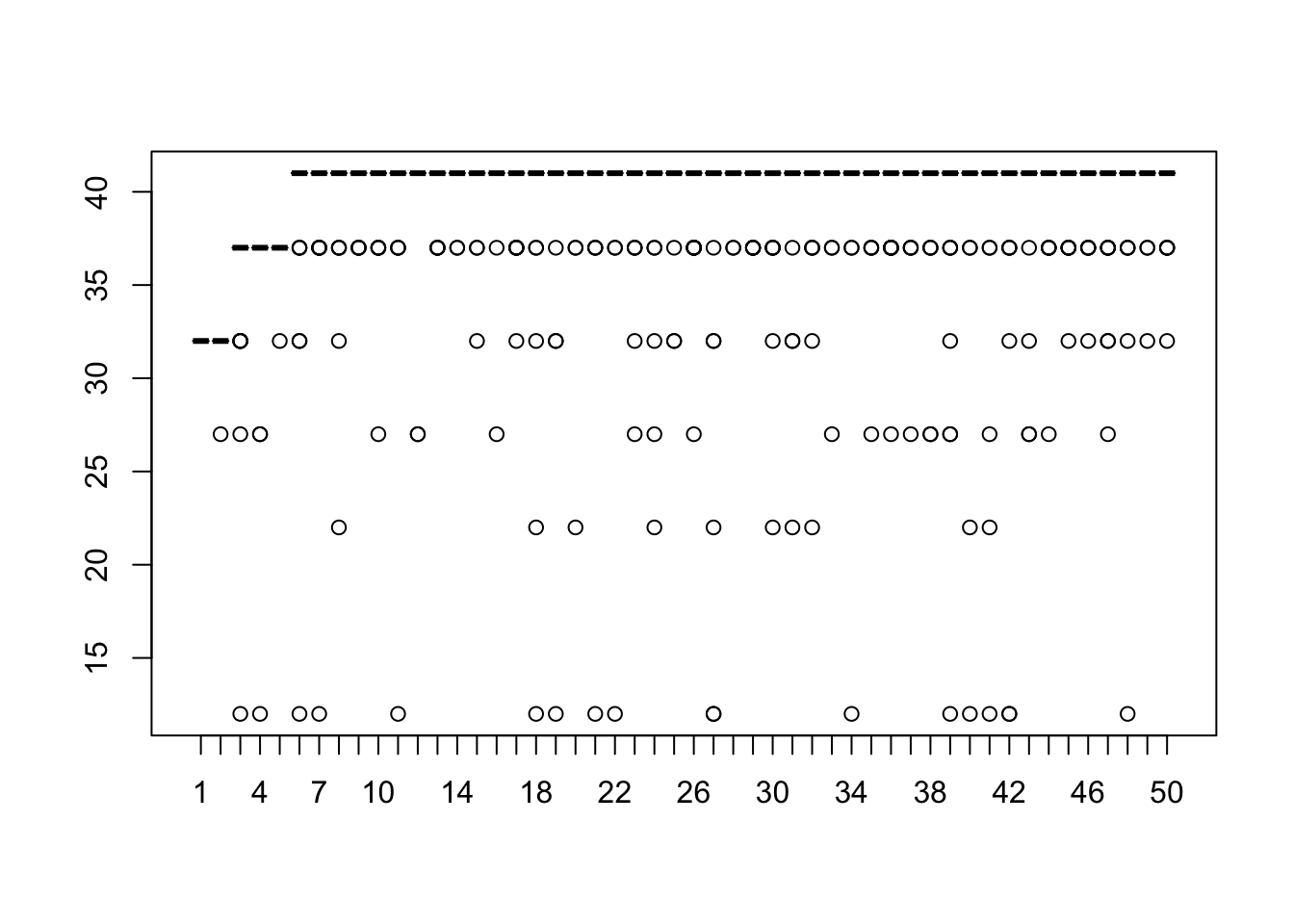
5.2 Salmon
Let’s go through using salmon to count reads directly from the trimmed fastq.gz files
5.2.1 Pseudomapping & Counting
DATA_DIR="/Users/$USER/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp"
SALMON_OUT_DIR="/Users/$USER/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon"
SALMON_INDEX_DIR="/Users/$USER/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1"
# make the analysis directory if it doesn't already exist
mkdir -p $SALMON_OUT_DIR
# activate the salmon environment
conda activate salmon
# loop through all of the fastq files
for fn in $DATA_DIR/*.fastq.gz;
do
samp=`basename ${fn}`
echo "Processing sample ${samp}"
# run salmon
salmon quant -i $SALMON_INDEX_DIR -l A \
-r ${fn} \
--useVBOpt \
-p 4 --validateMappings -o $SALMON_OUT_DIR/${samp}_quant
done
# combine all of the output files into a merged count matrix
salmon quantmerge --quants $SALMON_OUT_DIR/*_quant --column numreads -o $SALMON_OUT_DIR/salmon.gene_counts.merged.yeast.tsv
# remove the _mRNA from gene name
sed -i '' -E 's/^([^\t]+)_mRNA(\t|$)/\1\2/' $SALMON_OUT_DIR/salmon.gene_counts.merged.yeast.tsv
# we can also create a table of tpm values per gene by changing the --column flag
salmon quantmerge --quants $SALMON_OUT_DIR/*_quant --column tpm \
-o $SALMON_OUT_DIR/salmon.gene_tpm.merged.yeast.tsv
# remove the _mRNA from gene name
sed -i '' -E 's/^([^\t]+)_mRNA(\t|$)/\1\2/' $SALMON_OUT_DIR/salmon.gene_tpm.merged.yeast.tsv
conda deactivate## Processing sample YPS606_MSN24_ETOH_REP1_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_ETOH_REP1_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant/logs
## [2023-10-26 16:15:59.990] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:15:59.991] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:15:59.991] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:15:59.991] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:15:59.991] [jointLog] [info] parsing read library format
## -----------------------------------------
## | Loading contig table | Time = 6.3758 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 1.5446 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 191.33 us
## -----------------------------------------
## [2023-10-26 16:15:59.992] [jointLog] [info] There is 1 library.
## [2023-10-26 16:15:59.994] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:15:59.995] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading mphf table | Time = 11.646 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 35.129 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 5.7992 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 40.245 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 6.5275 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 579.67 us
## -----------------------------------------
## [2023-10-26 16:16:00.109] [jointLog] [info] done
## [2023-10-26 16:16:00.199] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:00.201] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:00.201] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:00.378] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:00.789] [jointLog] [info] Thread saw mini-batch with a maximum of 1.32% zero probability fragments
## [2023-10-26 16:16:00.808] [jointLog] [info] Thread saw mini-batch with a maximum of 1.40% zero probability fragments
## [2023-10-26 16:16:00.819] [jointLog] [info] Thread saw mini-batch with a maximum of 1.22% zero probability fragments
## [2023-10-26 16:16:00.824] [jointLog] [info] Thread saw mini-batch with a maximum of 1.32% zero probability fragments
## [2023-10-26 16:16:00.847] [jointLog] [info] Computed 5,478 rich equivalence classes for further processing
## [2023-10-26 16:16:00.847] [jointLog] [info] Counted 186,104 total reads in the equivalence classes
## [2023-10-26 16:16:00.854] [jointLog] [info] Number of mappings discarded because of alignment score : 31,126
## [2023-10-26 16:16:00.854] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 7,997
## [2023-10-26 16:16:00.854] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 6,248
## [2023-10-26 16:16:00.854] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:00.855] [jointLog] [warning] Only 186104 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:00.855] [jointLog] [info] Mapping rate = 79.7778%
##
## [2023-10-26 16:16:00.855] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:00.857] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:00.863] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:00.875] [jointLog] [info] iteration = 0 | max rel diff. = 2057.34
## [2023-10-26 16:16:02.144] [jointLog] [info] iteration = 100 | max rel diff. = 0.000215242
## [2023-10-26 16:16:02.145] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:02.145] [jointLog] [info] writing output
##
## Processing sample YPS606_MSN24_ETOH_REP2_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_ETOH_REP2_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:02.564] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:02.564] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:02.564] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:02.564] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:02.564] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:02.564] [jointLog] [info] There is 1 library.
## [2023-10-26 16:16:02.564] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:02.564] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig table | Time = 3.4287 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 129.58 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 35.583 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 5.9878 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 27.749 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.535 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 27.005 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.0361 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 76.584 us
## -----------------------------------------
## [2023-10-26 16:16:02.636] [jointLog] [info] done
## [2023-10-26 16:16:02.711] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:02.712] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:02.712] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:02.889] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:03.250] [jointLog] [info] Thread saw mini-batch with a maximum of 1.32% zero probability fragments
## [2023-10-26 16:16:03.274] [jointLog] [info] Thread saw mini-batch with a maximum of 1.22% zero probability fragments
## [2023-10-26 16:16:03.280] [jointLog] [info] Thread saw mini-batch with a maximum of 1.18% zero probability fragments
## [2023-10-26 16:16:03.281] [jointLog] [info] Thread saw mini-batch with a maximum of 1.40% zero probability fragments
## [2023-10-26 16:16:03.307] [jointLog] [info] Computed 5,293 rich equivalence classes for further processing
## [2023-10-26 16:16:03.307] [jointLog] [info] Counted 173,313 total reads in the equivalence classes
## [2023-10-26 16:16:03.314] [jointLog] [info] Number of mappings discarded because of alignment score : 25,173
## [2023-10-26 16:16:03.314] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 7,099
## [2023-10-26 16:16:03.314] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,772
## [2023-10-26 16:16:03.314] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:03.315] [jointLog] [warning] Only 173313 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:03.315] [jointLog] [info] Mapping rate = 80.3081%
##
## [2023-10-26 16:16:03.315] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:03.315] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:03.318] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:03.330] [jointLog] [info] iteration = 0 | max rel diff. = 1476.38
## [2023-10-26 16:16:04.568] [jointLog] [info] iteration = 100 | max rel diff. = 0.00017473
## [2023-10-26 16:16:04.568] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:04.568] [jointLog] [info] writing output
##
## Processing sample YPS606_MSN24_ETOH_REP3_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_ETOH_REP3_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:05.148] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:05.148] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:05.148] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:05.148] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:05.148] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:05.148] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 4.4033 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 100.25 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 33.875 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.0534 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## [2023-10-26 16:16:05.149] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:05.149] [jointLog] [info] Loading dense pufferfish index.
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 27.141 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.5127 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 27.572 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.2762 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 63.959 us
## -----------------------------------------
## [2023-10-26 16:16:05.221] [jointLog] [info] done
## [2023-10-26 16:16:05.289] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:05.290] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:05.290] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:05.463] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:05.726] [jointLog] [info] Thread saw mini-batch with a maximum of 1.58% zero probability fragments
## [2023-10-26 16:16:05.738] [jointLog] [info] Thread saw mini-batch with a maximum of 1.56% zero probability fragments
## [2023-10-26 16:16:05.742] [jointLog] [info] Thread saw mini-batch with a maximum of 1.52% zero probability fragments
## [2023-10-26 16:16:05.743] [jointLog] [info] Thread saw mini-batch with a maximum of 1.55% zero probability fragments
## [2023-10-26 16:16:05.766] [jointLog] [info] Computed 5,306 rich equivalence classes for further processing
## [2023-10-26 16:16:05.766] [jointLog] [info] Counted 158,068 total reads in the equivalence classes
## [2023-10-26 16:16:05.773] [jointLog] [info] Number of mappings discarded because of alignment score : 19,662
## [2023-10-26 16:16:05.773] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 6,467
## [2023-10-26 16:16:05.773] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 4,929
## [2023-10-26 16:16:05.773] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:05.773] [jointLog] [warning] Only 158068 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:05.773] [jointLog] [info] Mapping rate = 79.4008%
##
## [2023-10-26 16:16:05.773] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:05.774] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:05.777] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:05.788] [jointLog] [info] iteration = 0 | max rel diff. = 483.863
## [2023-10-26 16:16:07.056] [jointLog] [info] iteration = 100 | max rel diff. = 0.000307156
## [2023-10-26 16:16:07.056] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:07.056] [jointLog] [info] writing output
##
## Processing sample YPS606_MSN24_ETOH_REP4_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_ETOH_REP4_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:07.715] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:07.715] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:07.715] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:07.715] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:07.715] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:07.715] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.4127 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 97.084 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 30.875 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.0134 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## [2023-10-26 16:16:07.716] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:07.716] [jointLog] [info] Loading dense pufferfish index.
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 26.329 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.4595 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 27.063 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.1234 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 66.834 us
## -----------------------------------------
## [2023-10-26 16:16:07.786] [jointLog] [info] done
## [2023-10-26 16:16:07.853] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:07.854] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:07.854] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:08.029] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:08.297] [jointLog] [info] Thread saw mini-batch with a maximum of 1.26% zero probability fragments
## [2023-10-26 16:16:08.298] [jointLog] [info] Thread saw mini-batch with a maximum of 1.32% zero probability fragments
## [2023-10-26 16:16:08.300] [jointLog] [info] Thread saw mini-batch with a maximum of 1.26% zero probability fragments
## [2023-10-26 16:16:08.322] [jointLog] [info] Thread saw mini-batch with a maximum of 1.20% zero probability fragments
## [2023-10-26 16:16:08.342] [jointLog] [info] Computed 5,260 rich equivalence classes for further processing
## [2023-10-26 16:16:08.342] [jointLog] [info] Counted 165,617 total reads in the equivalence classes
## [2023-10-26 16:16:08.348] [jointLog] [info] Number of mappings discarded because of alignment score : 20,744
## [2023-10-26 16:16:08.348] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 6,497
## [2023-10-26 16:16:08.348] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,015
## [2023-10-26 16:16:08.348] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:08.348] [jointLog] [warning] Only 165617 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:08.348] [jointLog] [info] Mapping rate = 80.4779%
##
## [2023-10-26 16:16:08.348] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:08.348] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:08.351] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:08.361] [jointLog] [info] iteration = 0 | max rel diff. = 1859.21
## [2023-10-26 16:16:09.656] [jointLog] [info] iteration = 100 | max rel diff. = 0.000249168
## [2023-10-26 16:16:09.656] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:09.656] [jointLog] [info] writing output
##
## Processing sample YPS606_MSN24_MOCK_REP1_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_MOCK_REP1_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:10.282] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:10.282] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:10.282] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:10.282] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:10.282] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:10.282] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.5464 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 114.71 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 33.167 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 5.8997 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:10.282] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:10.282] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 27.302 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.4788 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 27.364 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.24 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 70.167 us
## -----------------------------------------
## [2023-10-26 16:16:10.354] [jointLog] [info] done
## [2023-10-26 16:16:10.421] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:10.422] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:10.422] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:10.591] [jointLog] [info] Automatically detected most likely library type as SR
##
## [2023-10-26 16:16:10.756] [jointLog] [info] Thread saw mini-batch with a maximum of 1.16% zero probability fragments
## [2023-10-26 16:16:10.765] [jointLog] [info] Thread saw mini-batch with a maximum of 1.20% zero probability fragments
## [2023-10-26 16:16:10.776] [jointLog] [info] Thread saw mini-batch with a maximum of 1.20% zero probability fragments
## [2023-10-26 16:16:10.780] [jointLog] [info] Thread saw mini-batch with a maximum of 1.18% zero probability fragments
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:10.798] [jointLog] [info] Computed 5,121 rich equivalence classes for further processing
## [2023-10-26 16:16:10.798] [jointLog] [info] Counted 130,772 total reads in the equivalence classes
## [2023-10-26 16:16:10.804] [jointLog] [info] Number of mappings discarded because of alignment score : 28,846
## [2023-10-26 16:16:10.804] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 6,943
## [2023-10-26 16:16:10.804] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,380
## [2023-10-26 16:16:10.804] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:10.804] [jointLog] [warning] Only 130772 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:10.804] [jointLog] [info] Mapping rate = 78.2714%
##
## [2023-10-26 16:16:10.804] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:10.805] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:10.807] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:10.816] [jointLog] [info] iteration = 0 | max rel diff. = 1687.17
## [2023-10-26 16:16:12.056] [jointLog] [info] iteration = 100 | max rel diff. = 0.000122859
## [2023-10-26 16:16:12.057] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:12.057] [jointLog] [info] writing output
##
## Processing sample YPS606_MSN24_MOCK_REP2_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_MOCK_REP2_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:12.865] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:12.865] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:12.865] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:12.865] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:12.865] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:12.865] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.6053 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 97.416 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 30.834 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 5.8202 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:12.865] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:12.865] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 26.288 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.4539 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 27.096 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.1037 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 71.625 us
## -----------------------------------------
## [2023-10-26 16:16:12.935] [jointLog] [info] done
## [2023-10-26 16:16:13.004] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:13.005] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:13.005] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:13.173] [jointLog] [info] Automatically detected most likely library type as SR
##
## [2023-10-26 16:16:13.346] [jointLog] [info] Thread saw mini-batch with a maximum of 1.10% zero probability fragments
## [2023-10-26 16:16:13.351] [jointLog] [info] Thread saw mini-batch with a maximum of 1.12% zero probability fragments
## [2023-10-26 16:16:13.364] [jointLog] [info] Thread saw mini-batch with a maximum of 1.12% zero probability fragments
## [2023-10-26 16:16:13.371] [jointLog] [info] Thread saw mini-batch with a maximum of 1.14% zero probability fragments
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:13.388] [jointLog] [info] Computed 5,107 rich equivalence classes for further processing
## [2023-10-26 16:16:13.388] [jointLog] [info] Counted 135,238 total reads in the equivalence classes
## [2023-10-26 16:16:13.394] [jointLog] [info] Number of mappings discarded because of alignment score : 19,440
## [2023-10-26 16:16:13.394] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 5,837
## [2023-10-26 16:16:13.394] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 4,792
## [2023-10-26 16:16:13.394] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:13.394] [jointLog] [warning] Only 135238 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:13.394] [jointLog] [info] Mapping rate = 79.667%
##
## [2023-10-26 16:16:13.394] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:13.394] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:13.397] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:13.405] [jointLog] [info] iteration = 0 | max rel diff. = 1365.23
## [2023-10-26 16:16:14.702] [jointLog] [info] iteration = 100 | max rel diff. = 0.00138672
## [2023-10-26 16:16:14.703] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:14.703] [jointLog] [info] writing output
##
## Processing sample YPS606_MSN24_MOCK_REP3_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_MOCK_REP3_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:15.601] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:15.601] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:15.601] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:15.601] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:15.601] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:15.601] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 4.5574 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 123.63 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 32.375 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.253 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## [2023-10-26 16:16:15.602] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:15.602] [jointLog] [info] Loading dense pufferfish index.
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 27.792 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.5702 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 28.717 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.0438 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 64.25 us
## -----------------------------------------
## [2023-10-26 16:16:15.676] [jointLog] [info] done
## [2023-10-26 16:16:15.750] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:15.751] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:15.751] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:15.968] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:16.453] [jointLog] [info] Thread saw mini-batch with a maximum of 1.52% zero probability fragments
## [2023-10-26 16:16:16.463] [jointLog] [info] Thread saw mini-batch with a maximum of 1.30% zero probability fragments
## [2023-10-26 16:16:16.487] [jointLog] [info] Thread saw mini-batch with a maximum of 1.58% zero probability fragments
## [2023-10-26 16:16:16.490] [jointLog] [info] Thread saw mini-batch with a maximum of 1.62% zero probability fragments
## [2023-10-26 16:16:16.519] [jointLog] [info] Computed 5,212 rich equivalence classes for further processing
## [2023-10-26 16:16:16.519] [jointLog] [info] Counted 161,111 total reads in the equivalence classes
## [2023-10-26 16:16:16.525] [jointLog] [info] Number of mappings discarded because of alignment score : 23,201
## [2023-10-26 16:16:16.525] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 7,784
## [2023-10-26 16:16:16.525] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 6,012
## [2023-10-26 16:16:16.525] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:16.525] [jointLog] [warning] Only 161111 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:16.525] [jointLog] [info] Mapping rate = 76.7192%
##
## [2023-10-26 16:16:16.525] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:16.526] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:16.529] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:16.540] [jointLog] [info] iteration = 0 | max rel diff. = 1651.79
## [2023-10-26 16:16:17.891] [jointLog] [info] iteration = 100 | max rel diff. = 0.000125455
## [2023-10-26 16:16:17.891] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:17.891] [jointLog] [info] writing output
##
## Processing sample YPS606_MSN24_MOCK_REP4_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_MSN24_MOCK_REP4_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant/logs
## -----------------------------------------
## | Loading contig table | Time = 4.4065 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 98.292 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 30.541 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.1712 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## [2023-10-26 16:16:18.767] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:18.767] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:18.767] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:18.767] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:18.767] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:18.767] [jointLog] [info] There is 1 library.
## [2023-10-26 16:16:18.767] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:18.767] [jointLog] [info] Loading dense pufferfish index.
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 26.763 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.5253 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 27.182 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.0114 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 85.584 us
## -----------------------------------------
## [2023-10-26 16:16:18.839] [jointLog] [info] done
## [2023-10-26 16:16:18.906] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:18.907] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:18.907] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:19.094] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:19.365] [jointLog] [info] Thread saw mini-batch with a maximum of 1.46% zero probability fragments
## [2023-10-26 16:16:19.373] [jointLog] [info] Thread saw mini-batch with a maximum of 1.46% zero probability fragments
## [2023-10-26 16:16:19.386] [jointLog] [info] Thread saw mini-batch with a maximum of 1.30% zero probability fragments
## [2023-10-26 16:16:19.388] [jointLog] [info] Thread saw mini-batch with a maximum of 1.28% zero probability fragments
## [2023-10-26 16:16:19.409] [jointLog] [info] Computed 5,243 rich equivalence classes for further processing
## [2023-10-26 16:16:19.409] [jointLog] [info] Counted 163,485 total reads in the equivalence classes
## [2023-10-26 16:16:19.415] [jointLog] [info] Number of mappings discarded because of alignment score : 20,601
## [2023-10-26 16:16:19.415] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 7,334
## [2023-10-26 16:16:19.415] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,778
## [2023-10-26 16:16:19.415] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:19.416] [jointLog] [warning] Only 163485 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:19.416] [jointLog] [info] Mapping rate = 78.4744%
##
## [2023-10-26 16:16:19.416] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:19.416] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:19.420] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:19.431] [jointLog] [info] iteration = 0 | max rel diff. = 1711.21
## [2023-10-26 16:16:20.762] [jointLog] [info] iteration = 100 | max rel diff. = 0.000307278
## [2023-10-26 16:16:20.762] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:20.762] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_ETOH_REP1_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_ETOH_REP1_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP1_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP1_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:21.344] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:21.344] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:21.344] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:21.344] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:21.344] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:21.344] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.4204 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 127.21 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 30.709 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 5.9205 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:21.344] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:21.344] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 26.265 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.4185 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 26.725 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.113 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 60.75 us
## -----------------------------------------
## [2023-10-26 16:16:21.414] [jointLog] [info] done
## [2023-10-26 16:16:21.480] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:21.481] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:21.481] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:21.654] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:21.872] [jointLog] [info] Thread saw mini-batch with a maximum of 1.40% zero probability fragments
## [2023-10-26 16:16:21.879] [jointLog] [info] Thread saw mini-batch with a maximum of 1.16% zero probability fragments
## [2023-10-26 16:16:21.882] [jointLog] [info] Thread saw mini-batch with a maximum of 1.36% zero probability fragments
## [2023-10-26 16:16:21.886] [jointLog] [info] Thread saw mini-batch with a maximum of 1.30% zero probability fragments
## [2023-10-26 16:16:21.905] [jointLog] [info] Computed 5,053 rich equivalence classes for further processing
## [2023-10-26 16:16:21.905] [jointLog] [info] Counted 149,230 total reads in the equivalence classes
## [2023-10-26 16:16:21.910] [jointLog] [info] Number of mappings discarded because of alignment score : 15,755
## [2023-10-26 16:16:21.910] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 5,253
## [2023-10-26 16:16:21.910] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 4,402
## [2023-10-26 16:16:21.910] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:21.911] [jointLog] [warning] Only 149230 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:21.911] [jointLog] [info] Mapping rate = 82.181%
##
## [2023-10-26 16:16:21.911] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:21.911] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:21.914] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:21.921] [jointLog] [info] iteration = 0 | max rel diff. = 1406.19
## [2023-10-26 16:16:23.115] [jointLog] [info] iteration = 100 | max rel diff. = 2.70713e-05
## [2023-10-26 16:16:23.115] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:23.115] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_ETOH_REP2_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_ETOH_REP2_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP2_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP2_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:23.931] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:23.931] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:23.931] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:23.931] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:23.931] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:23.931] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 5.807 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 134.04 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 34.542 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.0668 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## [2023-10-26 16:16:23.931] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:23.931] [jointLog] [info] Loading dense pufferfish index.
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 27.445 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.6328 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 28.627 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.1181 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 75.375 us
## -----------------------------------------
## [2023-10-26 16:16:24.007] [jointLog] [info] done
## [2023-10-26 16:16:24.076] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:24.077] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:24.077] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:24.267] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:24.607] [jointLog] [info] Thread saw mini-batch with a maximum of 1.36% zero probability fragments
## [2023-10-26 16:16:24.614] [jointLog] [info] Thread saw mini-batch with a maximum of 1.46% zero probability fragments
## [2023-10-26 16:16:24.617] [jointLog] [info] Thread saw mini-batch with a maximum of 1.12% zero probability fragments
## [2023-10-26 16:16:24.623] [jointLog] [info] Thread saw mini-batch with a maximum of 1.34% zero probability fragments
## [2023-10-26 16:16:24.649] [jointLog] [info] Computed 5,185 rich equivalence classes for further processing
## [2023-10-26 16:16:24.649] [jointLog] [info] Counted 163,064 total reads in the equivalence classes
## [2023-10-26 16:16:24.658] [jointLog] [info] Number of mappings discarded because of alignment score : 22,598
## [2023-10-26 16:16:24.658] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 6,365
## [2023-10-26 16:16:24.658] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,134
## [2023-10-26 16:16:24.658] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:24.658] [jointLog] [warning] Only 163064 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:24.658] [jointLog] [info] Mapping rate = 80.9046%
##
## [2023-10-26 16:16:24.658] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:24.660] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:24.664] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:24.678] [jointLog] [info] iteration = 0 | max rel diff. = 2044.58
## [2023-10-26 16:16:26.012] [jointLog] [info] iteration = 100 | max rel diff. = 0.000322112
## [2023-10-26 16:16:26.012] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:26.012] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_ETOH_REP3_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_ETOH_REP3_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP3_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP3_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:26.521] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:26.521] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:26.521] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:26.521] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:26.521] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:26.521] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.8293 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 130.42 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 32.208 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.6345 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:26.522] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:26.522] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 27.488 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.714 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 28.752 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 5.2932 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 84.25 us
## -----------------------------------------
## [2023-10-26 16:16:26.597] [jointLog] [info] done
## [2023-10-26 16:16:26.686] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:26.687] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:26.687] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:26.924] [jointLog] [info] Automatically detected most likely library type as SR
##
## [2023-10-26 16:16:27.376] [jointLog] [info] Thread saw mini-batch with a maximum of 1.78% zero probability fragments
## [2023-10-26 16:16:27.377] [jointLog] [info] Thread saw mini-batch with a maximum of 1.62% zero probability fragments
## [2023-10-26 16:16:27.389] [jointLog] [info] Thread saw mini-batch with a maximum of 1.72% zero probability fragments
## [2023-10-26 16:16:27.390] [jointLog] [info] Thread saw mini-batch with a maximum of 1.62% zero probability fragments
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:27.429] [jointLog] [info] Computed 5,301 rich equivalence classes for further processing
## [2023-10-26 16:16:27.429] [jointLog] [info] Counted 171,045 total reads in the equivalence classes
## [2023-10-26 16:16:27.435] [jointLog] [info] Number of mappings discarded because of alignment score : 18,641
## [2023-10-26 16:16:27.435] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 6,542
## [2023-10-26 16:16:27.435] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,138
## [2023-10-26 16:16:27.435] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:27.436] [jointLog] [warning] Only 171045 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:27.436] [jointLog] [info] Mapping rate = 79.6503%
##
## [2023-10-26 16:16:27.436] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:27.437] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:27.440] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:27.450] [jointLog] [info] iteration = 0 | max rel diff. = 2209.07
## [2023-10-26 16:16:28.651] [jointLog] [info] iteration = 100 | max rel diff. = 6.51043e-05
## [2023-10-26 16:16:28.651] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:28.651] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_ETOH_REP4_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_ETOH_REP4_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP4_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP4_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:29.683] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:29.683] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:29.683] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:29.683] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:29.683] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:29.683] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 4.7789 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 94.041 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 31.209 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 5.8783 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## [2023-10-26 16:16:29.684] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:29.684] [jointLog] [info] Loading dense pufferfish index.
## Inventory entries filled: 49
## -----------------------------------------
## | Loading contig boundaries | Time = 26.255 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.4236 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 26.623 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.1305 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 72.75 us
## -----------------------------------------
## [2023-10-26 16:16:29.754] [jointLog] [info] done
## [2023-10-26 16:16:29.827] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:29.828] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:29.828] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:29.996] [jointLog] [info] Automatically detected most likely library type as SR
##
## [2023-10-26 16:16:30.194] [jointLog] [info] Thread saw mini-batch with a maximum of 1.36% zero probability fragments
## [2023-10-26 16:16:30.195] [jointLog] [info] Thread saw mini-batch with a maximum of 1.26% zero probability fragments
## [2023-10-26 16:16:30.204] [jointLog] [info] Thread saw mini-batch with a maximum of 1.38% zero probability fragments
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:30.214] [jointLog] [info] Thread saw mini-batch with a maximum of 1.46% zero probability fragments
## [2023-10-26 16:16:30.230] [jointLog] [info] Computed 5,217 rich equivalence classes for further processing
## [2023-10-26 16:16:30.230] [jointLog] [info] Counted 151,376 total reads in the equivalence classes
## [2023-10-26 16:16:30.235] [jointLog] [info] Number of mappings discarded because of alignment score : 18,141
## [2023-10-26 16:16:30.235] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 5,851
## [2023-10-26 16:16:30.235] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 4,515
## [2023-10-26 16:16:30.235] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:30.236] [jointLog] [warning] Only 151376 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:30.236] [jointLog] [info] Mapping rate = 80.8119%
##
## [2023-10-26 16:16:30.236] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:30.236] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:30.238] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:30.246] [jointLog] [info] iteration = 0 | max rel diff. = 2134.58
## [2023-10-26 16:16:31.426] [jointLog] [info] iteration = 100 | max rel diff. = 0.000703782
## [2023-10-26 16:16:31.426] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:31.426] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_MOCK_REP1_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_MOCK_REP1_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP1_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP1_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:32.332] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:32.332] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:32.332] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:32.332] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:32.332] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:32.332] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.4363 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 108.08 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 32.042 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.0111 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:32.333] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:32.333] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 26.453 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.5232 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 26.941 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.0996 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 74.041 us
## -----------------------------------------
## [2023-10-26 16:16:32.403] [jointLog] [info] done
## [2023-10-26 16:16:32.470] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:32.471] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:32.471] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:32.635] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:32.901] [jointLog] [info] Thread saw mini-batch with a maximum of 1.38% zero probability fragments
## [2023-10-26 16:16:32.916] [jointLog] [info] Thread saw mini-batch with a maximum of 1.24% zero probability fragments
## [2023-10-26 16:16:32.917] [jointLog] [info] Thread saw mini-batch with a maximum of 1.30% zero probability fragments
## [2023-10-26 16:16:32.922] [jointLog] [info] Thread saw mini-batch with a maximum of 1.20% zero probability fragments
## [2023-10-26 16:16:32.939] [jointLog] [info] Computed 5,305 rich equivalence classes for further processing
## [2023-10-26 16:16:32.939] [jointLog] [info] Counted 177,055 total reads in the equivalence classes
## [2023-10-26 16:16:32.944] [jointLog] [info] Number of mappings discarded because of alignment score : 31,142
## [2023-10-26 16:16:32.944] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 8,597
## [2023-10-26 16:16:32.944] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 6,941
## [2023-10-26 16:16:32.944] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:32.944] [jointLog] [warning] Only 177055 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:32.944] [jointLog] [info] Mapping rate = 79.2054%
##
## [2023-10-26 16:16:32.944] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:32.944] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:32.947] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:32.954] [jointLog] [info] iteration = 0 | max rel diff. = 2091.83
## [2023-10-26 16:16:34.249] [jointLog] [info] iteration = 100 | max rel diff. = 0.000208332
## [2023-10-26 16:16:34.249] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:34.249] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_MOCK_REP2_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_MOCK_REP2_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP2_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP2_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:34.893] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:34.893] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:34.893] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:34.893] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:34.893] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:34.894] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 4.187 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 104.21 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 29.75 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 5.9423 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:34.894] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:34.894] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 26.304 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.5596 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 26.808 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.0102 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 65.5 us
## -----------------------------------------
## [2023-10-26 16:16:34.964] [jointLog] [info] done
## [2023-10-26 16:16:35.030] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:35.031] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:35.031] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:35.202] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:35.429] [jointLog] [info] Thread saw mini-batch with a maximum of 1.26% zero probability fragments
## [2023-10-26 16:16:35.429] [jointLog] [info] Thread saw mini-batch with a maximum of 1.32% zero probability fragments
## [2023-10-26 16:16:35.441] [jointLog] [info] Thread saw mini-batch with a maximum of 1.54% zero probability fragments
## [2023-10-26 16:16:35.451] [jointLog] [info] Thread saw mini-batch with a maximum of 1.46% zero probability fragments
## [2023-10-26 16:16:35.469] [jointLog] [info] Computed 5,175 rich equivalence classes for further processing
## [2023-10-26 16:16:35.469] [jointLog] [info] Counted 147,312 total reads in the equivalence classes
## [2023-10-26 16:16:35.475] [jointLog] [warning] 0.00160026% of fragments were shorter than the k used to build the index.
## If this fraction is too large, consider re-building the index with a smaller k.
## The minimum read size found was 27.
##
##
## [2023-10-26 16:16:35.475] [jointLog] [info] Number of mappings discarded because of alignment score : 24,562
## [2023-10-26 16:16:35.475] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 7,192
## [2023-10-26 16:16:35.475] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,915
## [2023-10-26 16:16:35.475] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:35.475] [jointLog] [warning] Only 147312 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:35.475] [jointLog] [info] Mapping rate = 78.5794%
##
## [2023-10-26 16:16:35.475] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:35.477] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:35.479] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:35.487] [jointLog] [info] iteration = 0 | max rel diff. = 1719.02
## [2023-10-26 16:16:36.895] [jointLog] [info] iteration = 100 | max rel diff. = 0.000465167
## [2023-10-26 16:16:36.895] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:36.895] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_MOCK_REP3_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_MOCK_REP3_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP3_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP3_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:37.481] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:37.481] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:37.481] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:37.481] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:37.481] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:37.481] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.5011 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 93.583 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 31.709 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.4904 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:37.481] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:37.481] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 27.659 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.6508 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 30.155 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 4.43 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 72.25 us
## -----------------------------------------
## [2023-10-26 16:16:37.557] [jointLog] [info] done
## [2023-10-26 16:16:37.631] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:37.632] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:37.632] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:37.835] [jointLog] [info] Automatically detected most likely library type as SR
##
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:38.275] [jointLog] [info] Thread saw mini-batch with a maximum of 1.58% zero probability fragments
## [2023-10-26 16:16:38.282] [jointLog] [info] Thread saw mini-batch with a maximum of 1.64% zero probability fragments
## [2023-10-26 16:16:38.289] [jointLog] [info] Thread saw mini-batch with a maximum of 1.58% zero probability fragments
## [2023-10-26 16:16:38.310] [jointLog] [info] Thread saw mini-batch with a maximum of 1.54% zero probability fragments
## [2023-10-26 16:16:38.336] [jointLog] [info] Computed 5,175 rich equivalence classes for further processing
## [2023-10-26 16:16:38.336] [jointLog] [info] Counted 173,912 total reads in the equivalence classes
## [2023-10-26 16:16:38.341] [jointLog] [info] Number of mappings discarded because of alignment score : 25,288
## [2023-10-26 16:16:38.341] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 8,408
## [2023-10-26 16:16:38.341] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 6,477
## [2023-10-26 16:16:38.341] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:38.342] [jointLog] [warning] Only 173912 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:38.342] [jointLog] [info] Mapping rate = 77.3743%
##
## [2023-10-26 16:16:38.342] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:38.342] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:38.345] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:38.355] [jointLog] [info] iteration = 0 | max rel diff. = 1677.12
## [2023-10-26 16:16:39.603] [jointLog] [info] iteration = 100 | max rel diff. = 7.7896e-05
## [2023-10-26 16:16:39.603] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:39.603] [jointLog] [info] writing output
##
## Processing sample YPS606_WT_MOCK_REP4_R1.fastq.gz
## Version Info: This is the most recent version of salmon.
## ### salmon (selective-alignment-based) v1.10.0
## ### [ program ] => salmon
## ### [ command ] => quant
## ### [ index ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Reference/index_salmon_Saccharomyces_cerevisiae.R64-1-1 }
## ### [ libType ] => { A }
## ### [ unmatedReads ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Trimmed_rfastp/YPS606_WT_MOCK_REP4_R1.fastq.gz }
## ### [ useVBOpt ] => { }
## ### [ threads ] => { 4 }
## ### [ validateMappings ] => { }
## ### [ output ] => { /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP4_R1.fastq.gz_quant }
## Logs will be written to /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP4_R1.fastq.gz_quant/logs
## [2023-10-26 16:16:40.066] [jointLog] [info] setting maxHashResizeThreads to 4
## [2023-10-26 16:16:40.066] [jointLog] [info] Fragment incompatibility prior below threshold. Incompatible fragments will be ignored.
## [2023-10-26 16:16:40.066] [jointLog] [info] Usage of --validateMappings implies use of minScoreFraction. Since not explicitly specified, it is being set to 0.65
## [2023-10-26 16:16:40.066] [jointLog] [info] Setting consensusSlack to selective-alignment default of 0.35.
## [2023-10-26 16:16:40.066] [jointLog] [info] parsing read library format
## [2023-10-26 16:16:40.066] [jointLog] [info] There is 1 library.
## -----------------------------------------
## | Loading contig table | Time = 3.6218 ms
## -----------------------------------------
## size = 25029
## -----------------------------------------
## | Loading contig offsets | Time = 155.04 us
## -----------------------------------------
## -----------------------------------------
## | Loading reference lengths | Time = 33.083 us
## -----------------------------------------
## -----------------------------------------
## | Loading mphf table | Time = 6.4004 ms
## -----------------------------------------
## size = 12321058
## Number of ones: 25028
## Number of ones per inventory item: 512
## Inventory entries filled: 49
## [2023-10-26 16:16:40.066] [jointLog] [info] Loading pufferfish index
## [2023-10-26 16:16:40.066] [jointLog] [info] Loading dense pufferfish index.
## -----------------------------------------
## | Loading contig boundaries | Time = 26.799 ms
## -----------------------------------------
## size = 12321058
## -----------------------------------------
## | Loading sequence | Time = 2.5237 ms
## -----------------------------------------
## size = 11570218
## -----------------------------------------
## | Loading positions | Time = 38.085 ms
## -----------------------------------------
## size = 20892357
## -----------------------------------------
## | Loading reference sequence | Time = 18.538 ms
## -----------------------------------------
## -----------------------------------------
## | Loading reference accumulative lengths | Time = 122.42 us
## -----------------------------------------
## [2023-10-26 16:16:40.163] [jointLog] [info] done
## [2023-10-26 16:16:40.274] [jointLog] [info] Index contained 6,588 targets
## [2023-10-26 16:16:40.275] [jointLog] [info] Number of decoys : 17
## [2023-10-26 16:16:40.275] [jointLog] [info] First decoy index : 6,571
##
##
##
##
## [2023-10-26 16:16:40.539] [jointLog] [info] Automatically detected most likely library type as SR
##
## [2023-10-26 16:16:40.929] [jointLog] [info] Thread saw mini-batch with a maximum of 1.08% zero probability fragments
## [2023-10-26 16:16:40.931] [jointLog] [info] Thread saw mini-batch with a maximum of 1.16% zero probability fragments
## [2023-10-26 16:16:40.944] [jointLog] [info] Thread saw mini-batch with a maximum of 1.38% zero probability fragments
## [2023-10-26 16:16:40.963] [jointLog] [info] Thread saw mini-batch with a maximum of 1.14% zero probability fragments
##
##
##
##
##
##
##
##
## [2023-10-26 16:16:41.005] [jointLog] [info] Computed 5,167 rich equivalence classes for further processing
## [2023-10-26 16:16:41.005] [jointLog] [info] Counted 163,571 total reads in the equivalence classes
## [2023-10-26 16:16:41.011] [jointLog] [info] Number of mappings discarded because of alignment score : 24,431
## [2023-10-26 16:16:41.011] [jointLog] [info] Number of fragments entirely discarded because of alignment score : 7,543
## [2023-10-26 16:16:41.011] [jointLog] [info] Number of fragments discarded because they are best-mapped to decoys : 5,763
## [2023-10-26 16:16:41.011] [jointLog] [info] Number of fragments discarded because they have only dovetail (discordant) mappings to valid targets : 0
## [2023-10-26 16:16:41.011] [jointLog] [warning] Only 163571 fragments were mapped, but the number of burn-in fragments was set to 5000000.
## The effective lengths have been computed using the observed mappings.
##
## [2023-10-26 16:16:41.011] [jointLog] [info] Mapping rate = 79.0714%
##
## [2023-10-26 16:16:41.011] [jointLog] [info] finished quantifyLibrary()
## [2023-10-26 16:16:41.012] [jointLog] [info] Starting optimizer
## [2023-10-26 16:16:41.015] [jointLog] [info] Marked 0 weighted equivalence classes as degenerate
## [2023-10-26 16:16:41.026] [jointLog] [info] iteration = 0 | max rel diff. = 1702.2
## [2023-10-26 16:16:42.401] [jointLog] [info] iteration = 100 | max rel diff. = 0.000337813
## [2023-10-26 16:16:42.401] [jointLog] [info] Finished optimizer
## [2023-10-26 16:16:42.401] [jointLog] [info] writing output
##
## Version Info: This is the most recent version of salmon.
## [2023-10-26 16:16:43.777] [mergeLog] [info] samples: [ /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP4_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP4_R1.fastq.gz_quant ]
## [2023-10-26 16:16:43.777] [mergeLog] [info] sample names : [ YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant, YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant, YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant, YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant, YPS606_WT_ETOH_REP1_R1.fastq.gz_quant, YPS606_WT_ETOH_REP2_R1.fastq.gz_quant, YPS606_WT_ETOH_REP3_R1.fastq.gz_quant, YPS606_WT_ETOH_REP4_R1.fastq.gz_quant, YPS606_WT_MOCK_REP1_R1.fastq.gz_quant, YPS606_WT_MOCK_REP2_R1.fastq.gz_quant, YPS606_WT_MOCK_REP3_R1.fastq.gz_quant, YPS606_WT_MOCK_REP4_R1.fastq.gz_quant ]
## [2023-10-26 16:16:43.777] [mergeLog] [info] output column : NUMREADS
## [2023-10-26 16:16:43.777] [mergeLog] [info] output file : /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/salmon.gene_counts.merged.yeast.tsv
## [2023-10-26 16:16:43.777] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.790] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.803] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.815] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.826] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.838] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.849] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.860] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.871] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.885] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.896] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.907] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP4_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.918] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.928] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.939] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:43.950] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP4_R1.fastq.gz_quant/quant.sf
## Version Info: This is the most recent version of salmon.
## [2023-10-26 16:16:44.401] [mergeLog] [info] samples: [ /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP4_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP1_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP2_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP3_R1.fastq.gz_quant, /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP4_R1.fastq.gz_quant ]
## [2023-10-26 16:16:44.401] [mergeLog] [info] sample names : [ YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant, YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant, YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant, YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant, YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant, YPS606_WT_ETOH_REP1_R1.fastq.gz_quant, YPS606_WT_ETOH_REP2_R1.fastq.gz_quant, YPS606_WT_ETOH_REP3_R1.fastq.gz_quant, YPS606_WT_ETOH_REP4_R1.fastq.gz_quant, YPS606_WT_MOCK_REP1_R1.fastq.gz_quant, YPS606_WT_MOCK_REP2_R1.fastq.gz_quant, YPS606_WT_MOCK_REP3_R1.fastq.gz_quant, YPS606_WT_MOCK_REP4_R1.fastq.gz_quant ]
## [2023-10-26 16:16:44.401] [mergeLog] [info] output column : TPM
## [2023-10-26 16:16:44.401] [mergeLog] [info] output file : /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/salmon.gene_tpm.merged.yeast.tsv
## [2023-10-26 16:16:44.401] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.413] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.424] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.436] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_ETOH_REP4_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.446] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.458] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.469] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.480] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_MSN24_MOCK_REP4_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.491] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.504] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.515] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.525] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_ETOH_REP4_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.538] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP1_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.549] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP2_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.560] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP3_R1.fastq.gz_quant/quant.sf
## [2023-10-26 16:16:44.570] [mergeLog] [info] Parsing /Users/clstacy/Desktop/Genomic_Data_Analysis/Data/Counts/Salmon/YPS606_WT_MOCK_REP4_R1.fastq.gz_quant/quant.sfThis script loops through each sample and invokes salmon using default mostly options. The -i argument tells salmon where to find the index -l A tells salmon that it should automatically determine the library type of the sequencing reads (e.g. stranded vs. unstranded etc.). The -r arguments tell salmon where to find the SE reads for this sample (notice, salmon will accept gzipped FASTQ files directly). Finally, the -p 4 argument tells salmon to make use of 4 threads and the -o argument specifies the directory where salmon’s quantification results should be written. The –useVBOpt flag sets to use variational Bayesian EM algorithm rather than the ‘standard EM’ to optimize abundance estimates (more accurate). Salmon exposes many different options to the user that enable extra features or modify default behavior. However, the purpose and behavior of all of those options is beyond the scope of this introductory tutorial. You can read about salmon’s many options in the documentation.
5.3 Questions
Identify which gene has the highest counts across all samples for both salmon and Rsubread outputs.
Redo the counting over the exons, rather than the genes (specify useMetaFeatures = FALSE) with RSubread. Use the bam files generated doing alignment reporting only unique reads, and call the featureCounts object fc.exon. Check the dimension of the counts slot to see how much larger it is.
What differences do you notice in the count values from Salmon vs Rsubread?
CHALLENGE: Download the full size fastq files from OneDrive & use Salmon to get the read counts on the non-subsampled files.
Be sure to knit this file into a pdf or html file once you’re finished.
System information for reproducibility:
R version 4.3.1 (2023-06-16)
Platform: aarch64-apple-darwin20 (64-bit)
locale: en_US.UTF-8||en_US.UTF-8||en_US.UTF-8||C||en_US.UTF-8||en_US.UTF-8
attached base packages: stats4, stats, graphics, grDevices, utils, datasets, methods and base
other attached packages: Rsubread(v.2.14.2), ShortRead(v.1.58.0), GenomicAlignments(v.1.36.0), SummarizedExperiment(v.1.30.2), MatrixGenerics(v.1.12.3), matrixStats(v.1.0.0), Rsamtools(v.2.16.0), GenomicRanges(v.1.52.1), Biostrings(v.2.68.1), GenomeInfoDb(v.1.36.4), XVector(v.0.40.0), BiocParallel(v.1.34.2), Rfastp(v.1.10.0), org.Sc.sgd.db(v.3.17.0), AnnotationDbi(v.1.62.2), IRanges(v.2.34.1), S4Vectors(v.0.38.2), Biobase(v.2.60.0), BiocGenerics(v.0.46.0), clusterProfiler(v.4.8.2), ggVennDiagram(v.1.2.3), tidytree(v.0.4.5), igraph(v.1.5.1), janitor(v.2.2.0), BiocManager(v.1.30.22), pander(v.0.6.5), knitr(v.1.44), here(v.1.0.1), lubridate(v.1.9.3), forcats(v.1.0.0), stringr(v.1.5.0), dplyr(v.1.1.3), purrr(v.1.0.2), readr(v.2.1.4), tidyr(v.1.3.0), tibble(v.3.2.1), ggplot2(v.3.4.4), tidyverse(v.2.0.0) and pacman(v.0.5.1)
loaded via a namespace (and not attached): RColorBrewer(v.1.1-3), rstudioapi(v.0.15.0), jsonlite(v.1.8.7), magrittr(v.2.0.3), farver(v.2.1.1), rmarkdown(v.2.25), fs(v.1.6.3), zlibbioc(v.1.46.0), vctrs(v.0.6.4), memoise(v.2.0.1), RCurl(v.1.98-1.12), ggtree(v.3.8.2), S4Arrays(v.1.0.6), htmltools(v.0.5.6.1), gridGraphics(v.0.5-1), sass(v.0.4.7), bslib(v.0.5.1), plyr(v.1.8.9), cachem(v.1.0.8), lifecycle(v.1.0.3), pkgconfig(v.2.0.3), Matrix(v.1.6-1.1), R6(v.2.5.1), fastmap(v.1.1.1), gson(v.0.1.0), GenomeInfoDbData(v.1.2.10), snakecase(v.0.11.1), digest(v.0.6.33), aplot(v.0.2.2), enrichplot(v.1.20.0), colorspace(v.2.1-0), patchwork(v.1.1.3), rprojroot(v.2.0.3), RSQLite(v.2.3.1), hwriter(v.1.3.2.1), fansi(v.1.0.5), timechange(v.0.2.0), abind(v.1.4-5), httr(v.1.4.7), polyclip(v.1.10-6), compiler(v.4.3.1), bit64(v.4.0.5), withr(v.2.5.1), downloader(v.0.4), viridis(v.0.6.4), DBI(v.1.1.3), ggforce(v.0.4.1), MASS(v.7.3-60), DelayedArray(v.0.26.7), rjson(v.0.2.21), HDO.db(v.0.99.1), tools(v.4.3.1), ape(v.5.7-1), scatterpie(v.0.2.1), glue(v.1.6.2), nlme(v.3.1-163), GOSemSim(v.2.26.1), grid(v.4.3.1), shadowtext(v.0.1.2), reshape2(v.1.4.4), fgsea(v.1.26.0), generics(v.0.1.3), gtable(v.0.3.4), tzdb(v.0.4.0), data.table(v.1.14.8), hms(v.1.1.3), tidygraph(v.1.2.3), utf8(v.1.2.3), ggrepel(v.0.9.4), pillar(v.1.9.0), vroom(v.1.6.4), yulab.utils(v.0.1.0), splines(v.4.3.1), tweenr(v.2.0.2), treeio(v.1.24.3), lattice(v.0.21-9), deldir(v.1.0-9), bit(v.4.0.5), tidyselect(v.1.2.0), GO.db(v.3.17.0), gridExtra(v.2.3), bookdown(v.0.36), xfun(v.0.40), graphlayouts(v.1.0.1), stringi(v.1.7.12), lazyeval(v.0.2.2), ggfun(v.0.1.3), yaml(v.2.3.7), evaluate(v.0.22), codetools(v.0.2-19), interp(v.1.1-4), ggraph(v.2.1.0), archive(v.1.1.5), qvalue(v.2.32.0), RVenn(v.1.1.0), ggplotify(v.0.1.2), cli(v.3.6.1), munsell(v.0.5.0), jquerylib(v.0.1.4), Rcpp(v.1.0.11), png(v.0.1-8), parallel(v.4.3.1), blob(v.1.2.4), jpeg(v.0.1-10), latticeExtra(v.0.6-30), DOSE(v.3.26.1), bitops(v.1.0-7), viridisLite(v.0.4.2), scales(v.1.2.1), crayon(v.1.5.2), rlang(v.1.1.1), cowplot(v.1.1.1), fastmatch(v.1.1-4) and KEGGREST(v.1.40.1)